Reconstructing evolutionary trees by maximum parsimony. Suppose we manage to sequence a particular gene across a whole bunch of different species. For concreteness, say there are n species, and the sequences are strings of length k over alphabet. How can we use this information to reconstruct the evolutionary history of these species?
Evolutionary history is commonly represented by a tree whose leaves are the different species, whose root is their common ancestor, and whose internal branches represent speciation events (that is, moments when a new species broke off from an existing one). Thus we need to find the following:
• An evolutionary tree with the given species at the leaves.
• For each internal node, a string of length K: the gene sequence for that particular ancestor.
For each possible tree T annotated with sequencesat each of its nodes , we can assign a score based on the principle of parsimony: fewer mutations are more likely.
localid="1659249441524"
Finding the highest-score tree is a difficult problem. Here we will consider just a small part of it: suppose we know the structure of the tree, and we want to fill in the sequences s(u) of the internal nodes u. Here’s an example with k=4 and n=5:
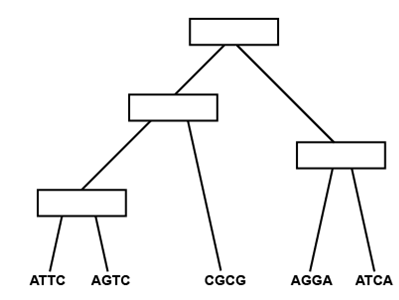
(a) In this particular example, there are several maximum parsimony reconstructions of the internal node sequences. Find one of them.
(b) Give an efficient (in terms of n and k ) algorithm for this task. (Hint: Even though the sequences might be long, you can do just one position at a time.)
